
This workshop provides a hands-on introduction to data preparation, genome assembly, and quality assessment. Participants will explore different assembly approaches and techniques for evaluating the accuracy and completeness of genome assemblies, helping attendees understand key metrics and statistical methods used to assess the quality of genomic data.
Tutorial Setup Instructions
Steps to prepare for the tutorial session:
- Login to Ceres Open OnDemand. For more information on login procedures for web-based SCINet access, see the SCINet access user guide.
- Open a command-line session by clicking on “Clusters” -> “Ceres Shell Access” on the top menu. This will open a new tab with a command-line session on Ceres’ login node.
-
Request resources on a compute node by running the following command.
srun --reservation=wk2_workshop -A scinet_workshop2 -t 05:00:00 -N 1 –c 8 --pty bash- For those accessing post-workshop: Remove
--reservation=wk2_workshopand change-A scinet_workshop2to your own project account. For additional information, see our SLURM guide.
- For those accessing post-workshop: Remove
-
Create a workshop working directory by running the following commands. Note: you do not have to edit the commands with your username as it will be determined by the $USER variable.
mkdir -p /90daydata/shared/$USER/genome_assembly cd /90daydata/shared/$USER/genome_assembly cp -r /project/scinet_workshop2/Bioinformatics_series/wk2_workshop/day1/ .
Data Preparation
by Viswanathan Satheesh and Sivanandan Chudalayandi
We have two data directories: 01_Data and 02_References
We start with checking the quality of data (Illumina and PacBio HiFi reads)
Illumina - FastQC
module load fastqc
mkdir 03_IlluminaQC
fastqc -o 03_IlluminaQC -t 2 01_Data/AT_Illumina_paired_*gz
Estimated Time: 1m 8s
-
Per base sequence quality
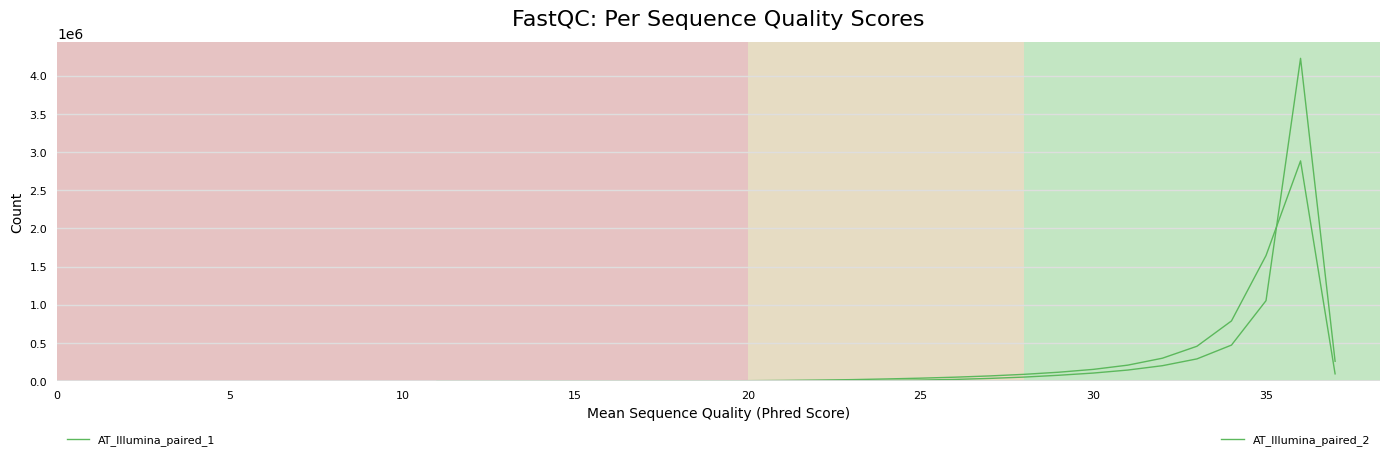
-
Per base sequence content

-
Overrepresented sequences
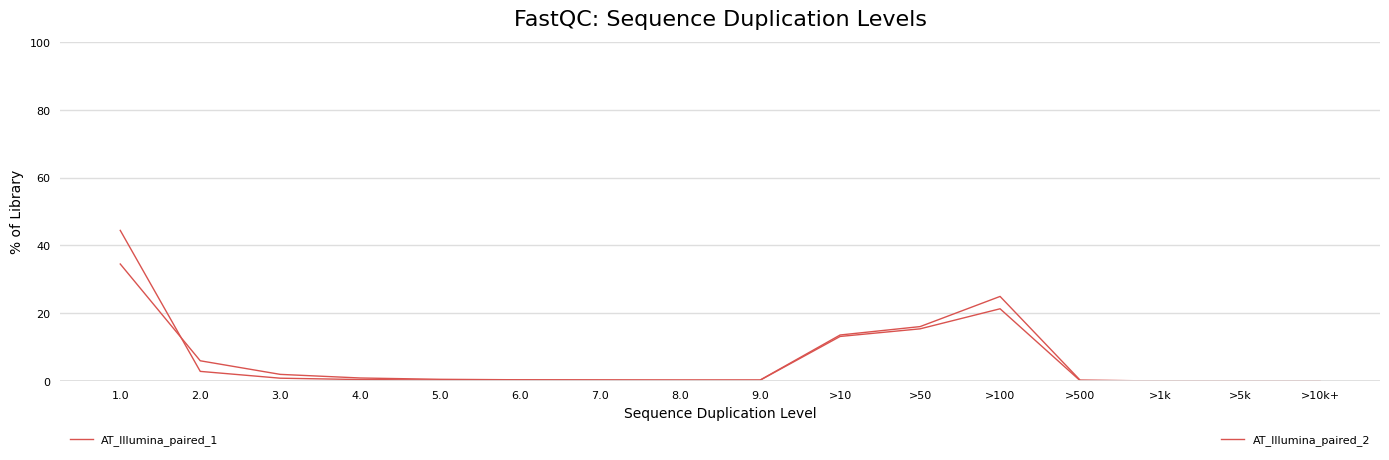
-
Adapter content
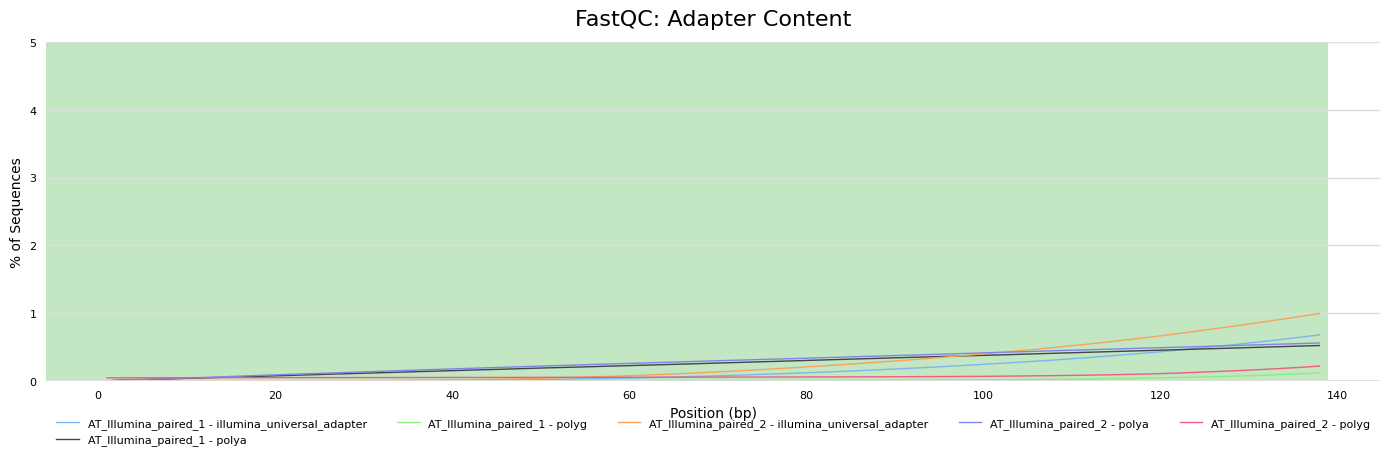
PacBio HiFi - nanoplot
module purge
module load miniconda
source activate /project/scinet_workshop2/Bioinformatics_series/nanoplot_conda/NP/
mkdir 04_NanoPlotQC
time NanoPlot --fastq 01_Data/mapped_reads.chr2.filtlong.fastq.gz -o 04_NanoPlotQC --threads 8
Estimated Time: 2m 17s
NanoPlot takes about 2 minute and 17 seconds to run.
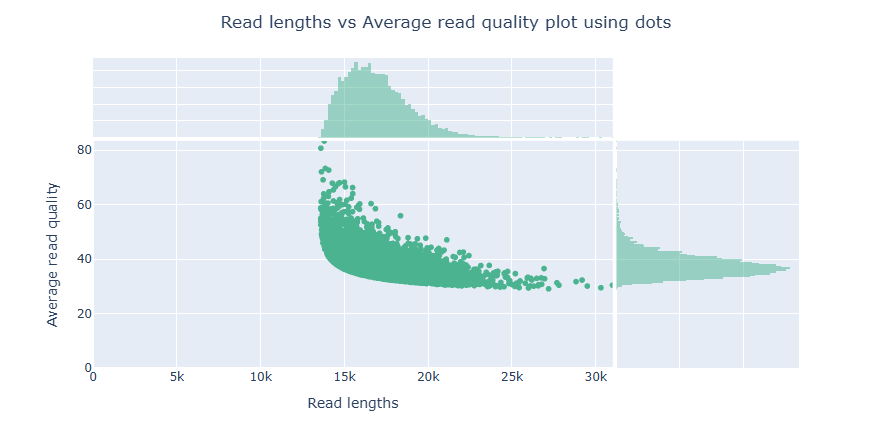
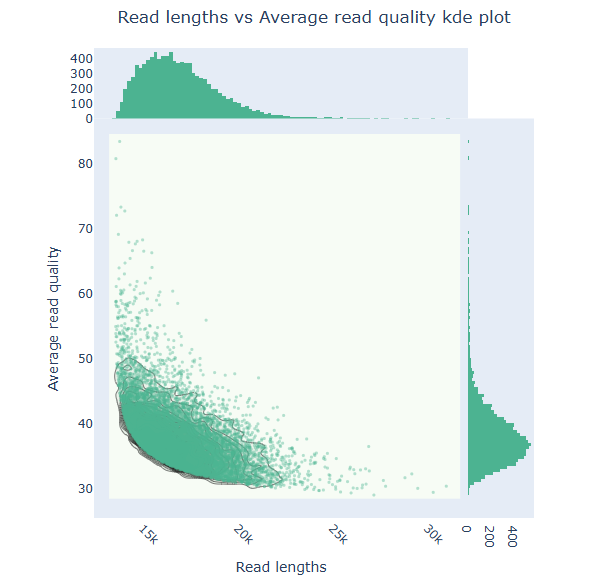
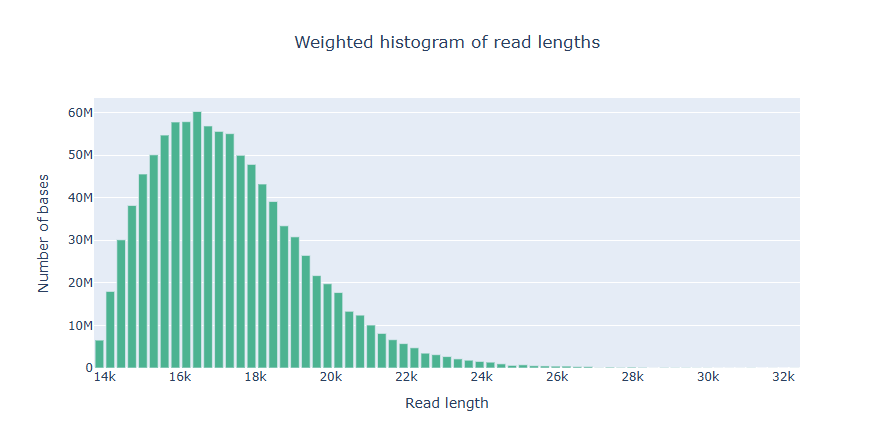
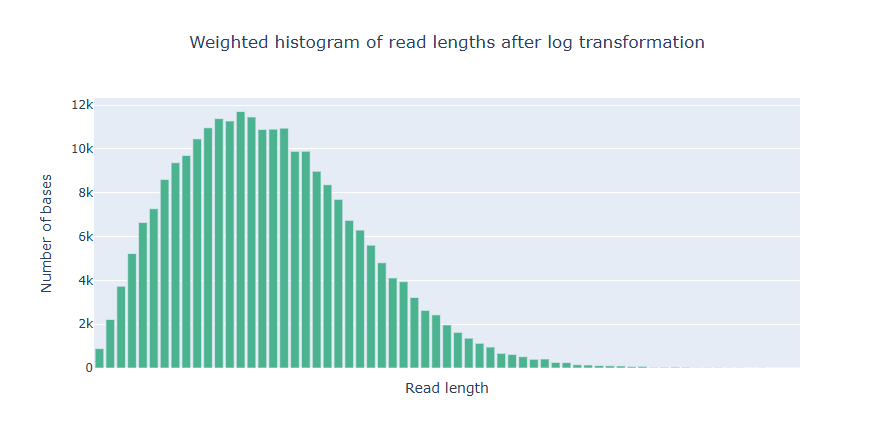
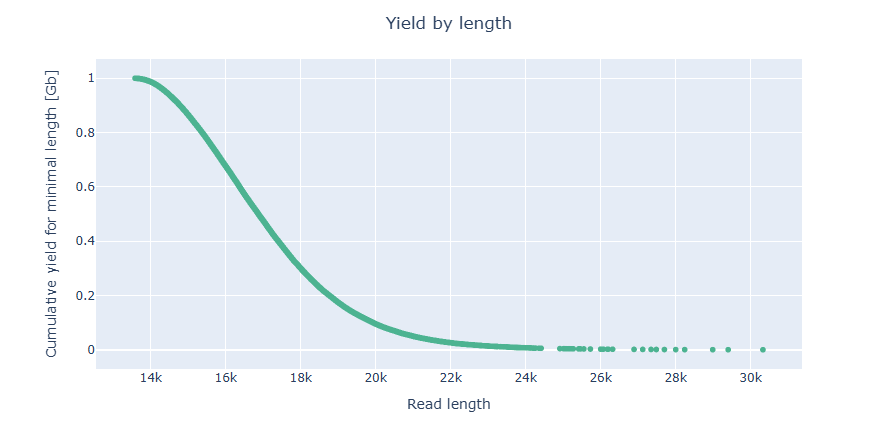
Predicting the genome size with Illumina reads with GenomeScope
module purge
module use /software/el9/spack/lmod/Core
module load jellyfish
mkdir 05_GenomeScope
unpigz 01_Data/AT*gz
time jellyfish count -m 21 -s 100M -t8 \
-C 01_Data/AT_Illumina_paired_* \
-o 05_GenomeScope/reads.jf
-m 21: 21-mers-s 10M: 10 million reads - this is the number of reads used to estimate the size of the genome-t 8: 8 threads
Takes about 1m 50 seconds to run.
time jellyfish histo -t 8 05_GenomeScope/reads.jf > 05_GenomeScope/reads.histo
jellyfish histois used to estimate the size of the genome using thereads.jffile. It outputs the size of the genome in thereads.histofile.
Takes about 20 seconds to run.
We can now copy the reads.histo to our local computer and upload it to GenomeScope
Interpretation of the images:
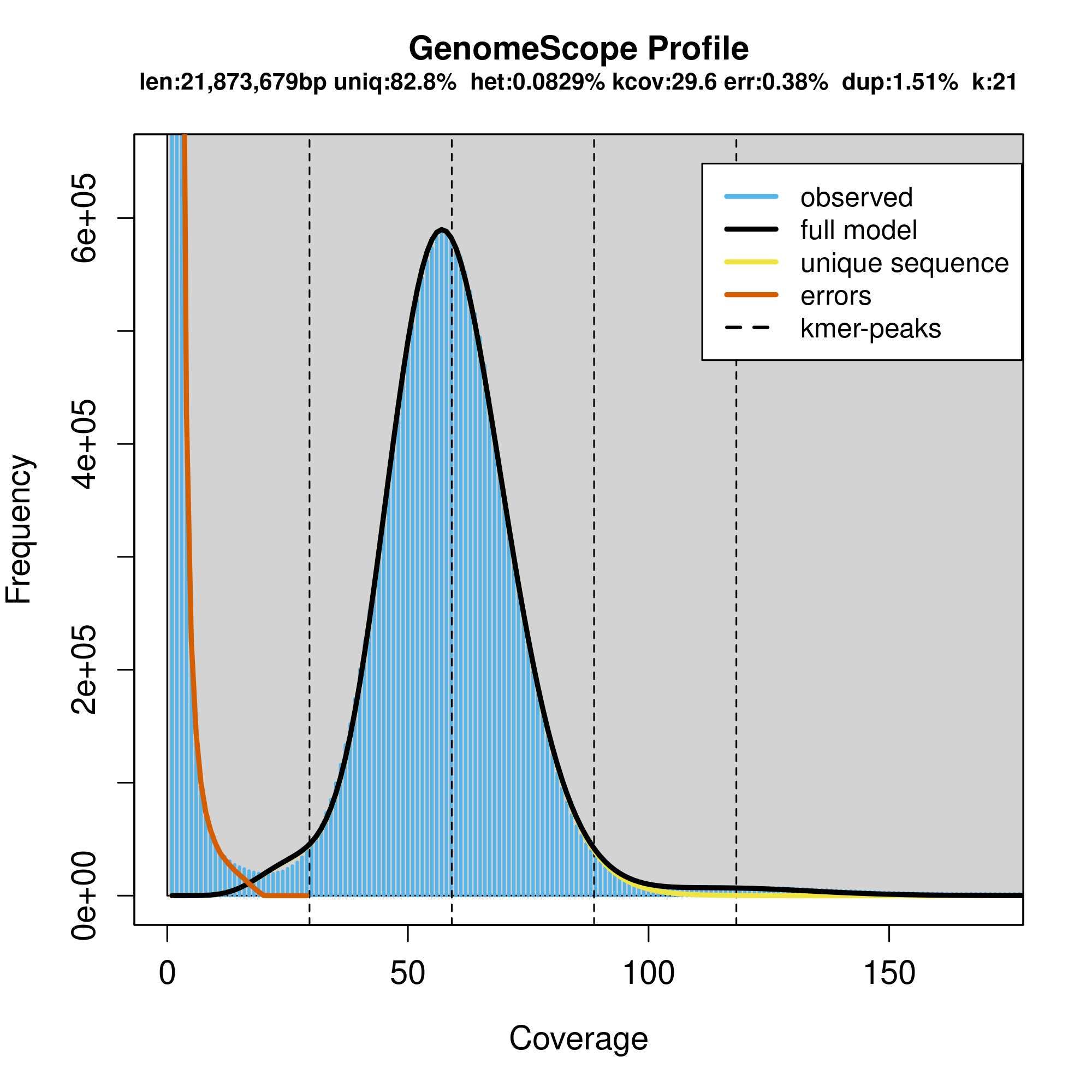
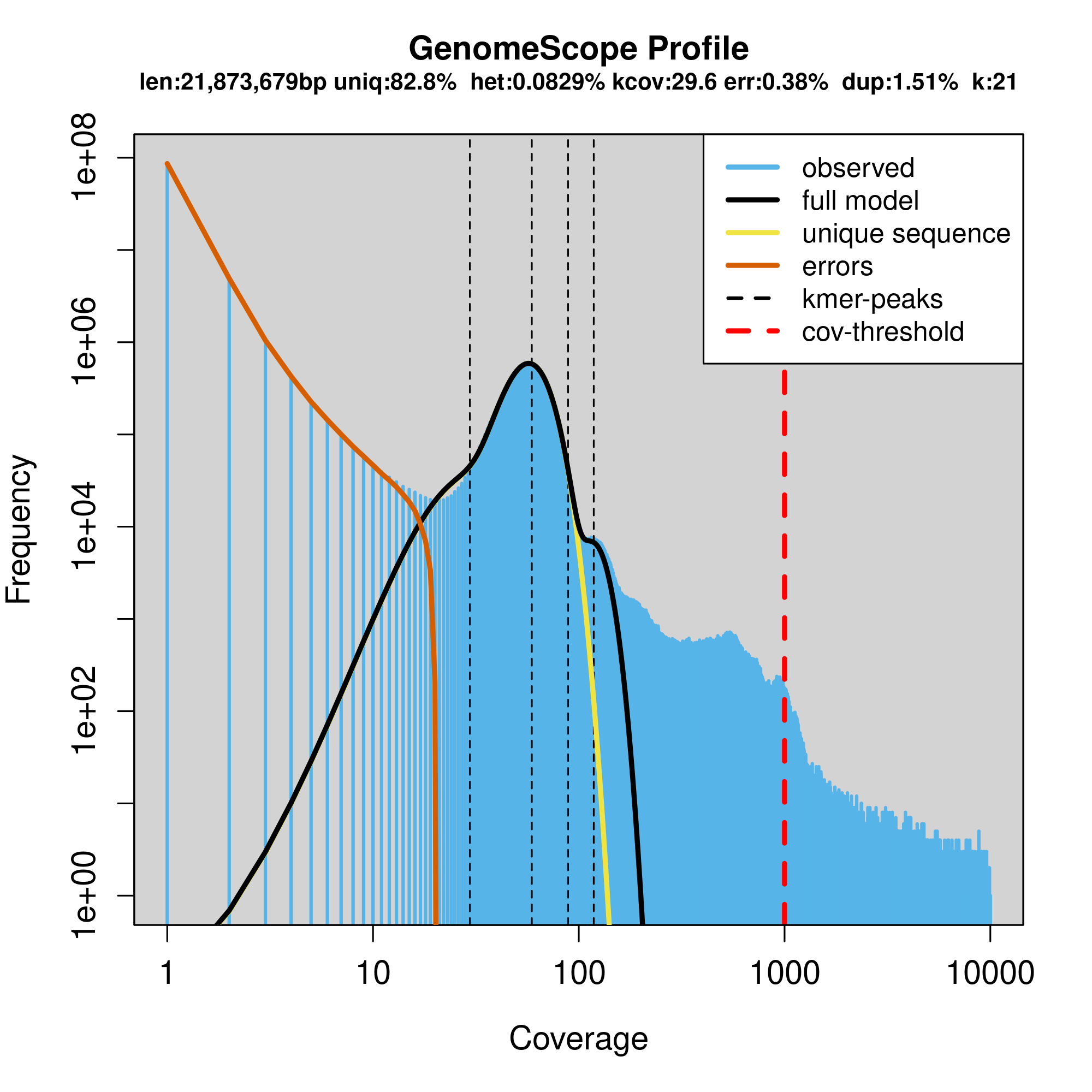
Common Elements in Both Figures
- Genome Size (len): Estimated genome size is 21,873,679 bp (~21 Mb).
- Unique Sequence (uniq): 82.8% of the genome is unique sequence.
- Heterozygosity (het): The heterozygosity rate is 0.0829%, indicating a very low level of heterozygosity.
- Coverage (kcov): Average k-mer coverage is 29.6.
- Error Rate (err): Estimated sequencing error rate is 0.38%.
- Duplication (dup): 1.51% of the genome is duplicated.
- k-mer size (k): k-mer length used for the analysis is 21.
Key Elements in the Plots
- X-Axis (Coverage): Represents the k-mer coverage. In the linear plot, it is shown on a linear scale, whereas in the logarithmic plot, it is shown on a logarithmic scale.
- Y-Axis (Frequency): Represents the frequency of k-mers at different coverage levels.
- Blue Bars (observed): Histogram of observed k-mer frequencies.
- Black Line (full model): Model fit to the observed k-mer frequencies.
- Yellow Line (unique sequence): Contribution of unique sequences to the k-mer frequencies.
- Orange Line (errors): Contribution of sequencing errors to the k-mer frequencies.
- Dashed Lines (kmer-peaks): Peaks corresponding to k-mer coverage of unique and repetitive sequences.
- Red Dashed Line (cov-threshold): A threshold to distinguish high-coverage k-mers, typically used to identify potential contaminant sequences or highly repetitive regions. This is set at a very high coverage level (around 1000).
Genome Assembly and Quality Assessment
This tutorial will guide you through the process of assembling a genome using HiFi reads and assessing its quality. We’ll be working with Arabidopsis thaliana chromosome 2 data as an example.
1. Genome Assembly with hifiasm
hifiasm is a fast and accurate haplotype-resolved assembler designed specifically for PacBio HiFi reads. It’s particularly effective because it can handle the high accuracy of HiFi reads while maintaining computational efficiency.
First, let’s create a directory for our assembly:
mkdir 06_Assembly
module purge
module load hifiasm
time hifiasm -o 06_Assembly/chr2_hifi.asm -t 8 -m 10 01_Data/mapped_reads.chr2.filtlong.fastq.gz
# Run hifiasm with the following parameters:
# -o: output prefix
# -t: number of threads (8 in this case)
# -m: minimum number of overlaps to keep a contig (10 in this case)
The assembly took approximately 3 minutes and 48 seconds to complete.
Converting Assembly Format from GFA to FASTA
The assembly output is in GFA (Graphical Fragment Assembly) format, which we need to convert to FASTA format for downstream analysis. GFA is a format that represents genome graphs, while FASTA is a more widely used format for sequence data.
# Extract sequences from GFA and convert to FASTA format
awk '/^S/ { print ">"$2; print $3 }' 06_Assembly/chr2_hifi.asm.bp.p_ctg.gfa > 06_Assembly/chr2_hifi.asm.bp.p_ctg.fa
Let’s check how many contigs we got from our assembly:
grep ">" -c 06_Assembly/chr2_hifi.asm.bp.p_ctg.fa
Our assembly resulted in 63 contigs.
Assembly Statistics
./new_Assemblathon.pl 06_Assembly/chr2_hifi.asm.bp.p_ctg.fa > 06_Assembly/chr2_AssemblyStats.txt
less 06_Assembly/chr2_AssemblyStats.txt
2. Quality Assessment
BUSCO Analysis with compleasm
BUSCO (Benchmarking Universal Single-Copy Orthologs) is a crucial tool for assessing genome assembly completeness. It searches for highly conserved genes that should be present in all species of a given lineage. The presence, absence, or fragmentation of these genes gives us insight into the quality of our assembly.
Key BUSCO metrics:
- Complete (S): Single-copy complete genes
- Duplicated (D): Complete genes present multiple times
- Fragmented (F): Partially assembled genes
- Missing (M): Genes not found in the assembly
- Total (N): Total number of genes in the BUSCO dataset
We’ll use compleasm, a faster implementation of BUSCO, with the embryophyta_odb12 database which is specific for plants:
module purge
module load miniconda
source activate /project/scinet_workshop2/Bioinformatics_series/Compleasm_conda/
time compleasm run -t 8 -l eukaryota -L 01_Data/busco/eukaryota_odb12 -a 06_Assembly/chr2_hifi.asm.bp.p_ctg.fa -o 07_Compleasm
This takes about 5 mins to run.
Understanding the BUSCO Results
Our assembly of chromosome 2 shows:
- 20.93% (27) complete single-copy genes
- 0.00% (0) duplicated genes
- 2.33% (3) fragmented genes
- 76.47% (99) missing genes
- Total: 129 genes assessed
These numbers are expected to be low since we’re only looking at one chromosome rather than the complete genome. For comparison, let’s look at the metrics for the complete Arabidopsis genome:
3. K-mer Based Quality Assessment
K-mer analysis is another powerful method for evaluating genome assembly quality. A k-mer is a substring of length k from a DNA sequence. By analyzing the distribution and frequency of k-mers in both our raw reads and assembly, we can assess assembly completeness and accuracy.
Step 1: Determining Optimal K-mer Size
First, we need to determine the optimal k-mer length for our analysis. This is important because:
- Too small k-mers may lead to random matches
- Too large k-mers may miss legitimate matches
- The optimal size depends on genome size and error rates
module purge
module load merqury
# Calculate optimal k-mer size based on genome size (19MB)
best_k.sh 19000000
Output:
genome: 19000000 tolerable collision rate: 0.001 17.0719
The script suggests a k-mer size of 17 based on our genome size of 19Mb and a tolerable collision rate of 0.001.
Step 2: K-mer Analysis with Merqury
Merqury is a tool that evaluates genome assemblies using k-mer frequencies. It helps us understand:
- Assembly completeness
- Base-level accuracy
- Potential misassemblies
Let’s run the analysis:
# Count k-mers in the input reads
time meryl k=17 count output 08_AT_HiFi.meryl 01_Data/mapped_reads.chr2.filtlong.fastq.gz
# Create output directory and run Merqury
mkdir 09_Merqury_Output_HiFi && cd 09_Merqury_Output_HiFi
merqury.sh ../08_AT_HiFi.meryl \
../06_Assembly/chr2_hifi.asm.bp.p_ctg.fa merqury_out
Step 3: Interpreting Merqury Results
Quality Value (QV) Statistics
Let’s examine the assembly quality values:
cat merqury_out.qv
chr2_hifi.asm.bp.p_ctg 40 28858272 70.8866 8.15344e-08
These metrics tell us:
- Assembly Name: chr2_hifi.asm.bp.p_ctg
- Unique K-mers: 40 k-mers found only in the assembly
- Total K-mers: 28,858,272 k-mers found in both assembly and reads
- Quality Value (QV): 70.89 - this is excellent! QV is a logarithmic measure of error probability
- Error Rate: 8.15e-08 - extremely low, indicating very high accuracy
Completeness Assessment
Let’s check the assembly completeness:
cat merqury_out.completeness.stats
chr2_hifi.asm.bp.p_ctg all 18214072 18235904 99.8803
These statistics show:
- Covered Bases: 18,214,072 bases are supported by k-mers from our input reads
- Total Bases: 18,235,904 bases in our assembly
- Completeness: 99.88% - this is exceptional! It means almost every base in our assembly is supported by the input reads
Exercises
-
Download the saccharmycetes busco lineage and run compleasm on the yeast genome assembly in
00_Miscellaneous_Data/yeast.fasta
Hint: checkcompleasm download -h -
Use the yeast nanopore reads
00_Miscellaneous_Data/SRR18210286_filtlong_500Mb.fastq.gzto get the merqury qv of the yeast genome assembly00_Miscellaneous_Data/yeast.fasta.
Hint: The genome size of yeast is approximately 12 MB.
Workshop Materials
Workshop materials and recordings available for USDA employees at the links below